������C��Ҏ�P�϶������IJ���֮̎���M����
�����Խ�������P�϶���������A���������b���M����һ�����о����M��ʹ�����r�g���s�̣���ᘌ����ָʾ����횬F�ìF���ҵζ��K�c�����Д�IJ��㣬�x�����Ӌ������ָʾ���Д�ζ��ĽK�c��ʹ�y���Y���Ĝʴ_����ߡ���������������ĵ����|�����y���������H�������㣬������Ⱦ�p�٣��Y�����Ӝʴ_��
����1 ԇ�����c����
����1.1 ԇ�x����ԇ��
����1.1.1 �x�� �P�϶����x�����ζ��ܡ�����ƿ ��100 mL�����F��ƿ ��100 mL��250 mL�� �����t��©�� ���������ܡ�ͨ�L�������Ӌ��늠t��
����1.1.2 ԇ���G���ۡ���Ȟ� 0.01 mol/L�}��˜���Һ�������ᡢ�w�e�֔� 1%��������Һ���|���֔� 10%�����~��Һ������⛡��|���֔� 25%�������c��Һ�����tһ�μ��{���ָʾ����30%�^�����䡢ϡ�A��Һ�������@�������@��
����1.2 ���E
����1.2.1 �����b�õĽM�b
������ԭ���P�϶�������Ҫ�̶��ĄP�ϟ�ƿ�Q�ɲ���Ҫ�̶��� 250 mL ���F��ƿ��ͬ�r��ƿ�ڵ���һ©����©���i픲��ò��������B�ӣ��������Ì����c��ʢϡ�A��Һ�Ĵ�������B��
����1.2.2 �����Qȡ
���������Ʒ 0.2~0.3 g,�������� 250 mL ���F��ƿ�У��ټ����|���֔� 10%�������~ 5 mL ��0.30 g ����⛼� 8.00 mL �����ᣬͬ�r���հ��Ռ��u���ƿ���ډ|���F�z�W��늠t�ϣ���ƿ�ڵ���һ©����©���i픲��ò��������B�ӣ��������Ì����c��ʢϡ�A��Һ�Ĵ�������B���b�ð��b�ú���ͨ�L���У����_늠t������ƿֱ�Ӽӟᣬ�M�������������С���S�r�{����Ĵ�С��ʹ�a���ğ��ⱻ�AҺ��ȫ���գ���������ȫ��̿������ĭ��ȫֹͣ��ӏ��������S�r�D�ӟ�ƿ��ʹƿ���ϵă�����ȫ������������Һ�У�������Һ���������Ұף�ȡ�´��䡣�� 10 mL���sˮ��ƿ�ڼ����F��ƿ�ȣ��D�� 100mL ����ƿ�ȣ������sˮ�_ϴ���Σ�ϴҺ�ϲ�������ƿ�У�����s��ϡ����̶ȣ����á�
����1.2.3 �y��������sˮ �����sˮ�l��ƿ�У���
������ 100 mL �F��ƿ�ȼ��� 15 mL �w�e�֔��� 1%��������Һ�������������£���ʹ�ܿڽ�������ȣ��A�o�Ś�ڣ�ȡ10 mL ��Ʒϡ�Һ��հ�Һ���M�ӿ�ע�뷴���ҡ���5 mL ���sˮ�_ϴ�ӿڣ�����Ͳ��ȡ 5 mL �|���֔�25%NaOH,Ѹ�ٵ����M�ӿڣ����������ã���ˮ���M�ӿڣ��Է����ݳ����ĵ� 1 ����Һ�����_ʼӋ�r�����s 3 min,�Ƅ�����ƿ��ʹ����Һ���x�_�����ܿڣ������s 1 min,Ȼ�����������sˮ�_ϴ�������¶ˣ��Ķ����C�����x������ȫ���s���ա����Ƿ���ȫ���s���������� pH ԇ��ԇ��s��Һ�Ƿ��A�Զ��_����ȡ������ƿ�������Ӌ�ζ��� pH ֵ 5.1��ȡ���ָʾ��׃ɫ�c��ֵ����ӛ����������w�e��mL�����^�m�A�o�Ś�ڣ������M������ʹ���sˮ���뷴���ң���o�M����Ƥ�ܣ��Ԕ�^����Դ���@�r�������еďUҺ���Ԅ���������˷��͛_ϴ�Ƀ������ң����Ś��y���_��ʹ����������еďUҺ�ų���
����2 �Y���c����
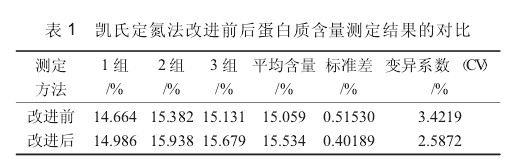
����2.1 �����|�����y���Y�����ȷքe���ø��Mǰ����ĄP�϶��������y�� 3 �M�G����ƽ��ԇ���еĵ����|�������P�϶��������Mǰ���|�����y���Y���Č���Ҋ�� 1.�ɱ� 1 ���Կ������P�϶���������ǰ��ĵ����|�����y���Y������һ�£������M��Ę˜ʲ��׃��ϵ��׃С���f�����M��Ĝy���������и��ߵľ��ܶȡ�
����1.2 �ζ��K�c�ʴ_�Ⱥ;��ܶȵı��^�������Ӌ��ָʾ�� 2 �N�����Д�ζ��K�c�Ĝʴ_�Ⱥ;��ܶȡ�
������ͬ�ζ����ľ��ܶȱ��^Ҋ�� 2,��ͬ�ζ����Ĝʴ_�ȱ��^Ҋ�� 3.�ɱ� 2 �ͱ� 3 ���Կ�����2 �N�����y���Y����������ܶȺ͜ʴ_�ȶ����Ϸ���Ҫ���ɱ� 5 ���Կ��������M��������r�g�s�̣�С��ԭ�����r�g�� 1/3,�@���������ڸ��M����ܟ���e׃�ӿ��������ٶȣ�������a���Ě��w�ֱ�ϡ�AҺ���գ����p�����к����w���ų���
����3 �YՓ
������1�� ͨ�^ȫ�ԄӶ����x���M�ĄP�϶������������b�ÿ��Կs�������r�g���Ķ���ʡ�������YԴ��
������2�� ���M��Ĝy�����������@�p���� CO2,SO2,SO3�Ț��w���ŷ��������o�˭h�������������w������
������3�� �����Ӌ����ָʾ����ʹ�Y�����Ӿ��ܺ͜ʴ_��


